

Хвороба Штаргардта
29.01.2019Хвороба Штаргардта – спадкове захворювання сітківки, яке проявляється дистрофічними змінами її макулярної зони і призводить до втрати центрального зору.
Хвороба Штаргардта
Інша назва хвороби Штаргардта – ювенільний макулярна дегенерація – відображає суть захворювання: воно починається в юному (ювенильном) віці і характеризується ураженням макули – рецепторного апарату зорового аналізатора. Захворювання описав німецький офтальмолог Карл Штаргардта на початку ХХ століття як вроджене ураження макулярної області очі, яке передавалося у спадок в одній сім’ї.
Типові Офтальмоскопически ознаки хвороби Штаргардта поліморфні:
- «атрофія хоріоідеі»,
- «бичаче око»,
- «бита (ковані) бронза».
Патогенетичне назва патології – «желтопятністая абіотрофія сітківки» – відображає зміни в області очного дна.
У 1997 році генетики виявили мутацію гена АВСR, що викликає порушення вироблення білка, який повинен переносити енергію фоторецепторних клітинам. Неповноцінність переносника АТФ призводить до загибелі фоторецепторів сітківки очей. Різні види спадково обумовленої макулярної дистрофії зустрічаються в 50% випадків патології очей. З них хвороба Штаргардта становить близько 7%. Нозологічна форма діагностується з частотою 1: 10000 та характеризується прогресуючим перебігом.
Двостороння патологія очей починається в молодому віці (від 6 до 21 років) і призводить до тяжких наслідків, аж до повної втрати зору. Захворювання має соціальну значимість, тому що призводить до інвалідності в молодому віці.
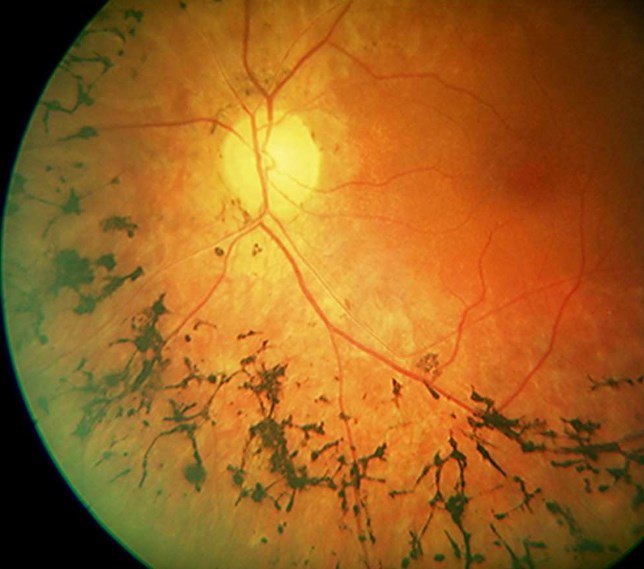
Абіотрофія сітківки
Дебют захворювання припадає на дитячий або юнацький вік. У пацієнтів відзначаються центральні скотоми і порушення колірного зору. Прогресування хвороби Штаргардта призводить до повної сліпоти.
Діагностика проводиться за допомогою:
- офтальмоскопії,
- флуоресцентної ангіографії
- ЕФД сітківки.
Для лікування застосовується ін’єкційна терапія (вітаміни, антиоксиданти, Ангіопротектори), фізіотерапія, проводяться реваскулярізірующіх операції, розробляється методика аутологічної тканинної терапії.
Причини розвитку хвороби
Спадкування не залежить від статевої приналежності пацієнта і батьків. Патологія передається переважно по аутосомно-рецесивним типом, тобто наслідування патології не пов’язано з підлогою (аутосомное – пов’язано з нестатевими хромосомами) і не завжди передається майбутньому поколінню (рецесивний шлях успадкування). За останніми даними лікарів-генетиків, патологія гена може передаватися і за домінантним типом. При домінантному типі успадкування дефектів гена – контролера синтезу білка-транспортера АТФ – захворювання протікає легше і нечасто призводить до інвалідності. Більшість рецепторних клітин макули (верхівки) жовтої плями очного дна функціонують. У пацієнтів з домінантним типом успадкування хвороба протікає з мінімумом проявів. Хворі зберігають працездатність і можуть навіть водити автотранспорт.
Основна причина дегенерації клітин макули полягає в тому, що вони страждають від дефіциту енергії. Дефект гена призводить до синтезу неповноцінного білка, що транспортує молекули АТФ через мембрану клітин жовтої плями – центру сітківки ока, в якому фокусується графічне і кольорове зображення. В області жовтої плями немає кровоносних судин. Харчування клітин-колб здійснюється за рахунок білків-переносників АТФ з прилеглої судинної оболонки (хориоидеи). Білки переносять через мембрану всередину клітин-колб молекули АТФ.
Хвороба Штаргардта – причини, симптоми
У нормальних умовах родопсин фоторецепторів поглинає фотон світла, трансформуючись в транс-ретиналь і опсин. Потім транс-ретиналь під дією енергії АТФ, що хтось принесе білки-переносники, перетворюється в ретиналь, який з’єднується з опсин. Так відновлюється родопсин. При спадкової мутації гена утворюється неповноцінний білок-переносник. В результаті порушується відновлення родопсину і накопичується транс-ретиналь. Він перетворюється в липофусцин і діє токсично на клітини-колбочки.
Класифікація хвороби
Види захворювання залежать від поширеності зони ураження жовтої плями. В офтальмології розрізняють наступні форми хвороби Штаргардта:

- центральну,
- періцентральная,
- центроперіферіческую (змішану).
При центральній формі уражаються клітини в центрі жовтої плями. Це виражається в випаданні центрального зору. У хворого з’являється центральна скотома (від гр. «Скотос» – темрява). Випадає з поля зору центральна зона. Хворий бачить зображення з темною плямою в точці фіксації погляду.
Періцентральная форма характеризується появою скотоми в стороні від точки фіксації. Людина здатна фокусувати погляд, але зазначає випадання в однієї зі сторін від центра поля зору у вигляді півмісяця. Згодом скотома набуває вигляду темного кільця. Центроперіферіческая форма починається з центру і стрімко поширюється до периферії. Темна пляма розростається і повністю перекриває поле зору.
Симптоми хвороби
Прояви захворювання починаються у віці 6-7 років. У всіх пацієнтів, незалежно від типу успадкування, спостерігаються центральні скотоми. При сприятливому перебігу скотоми відносні: пацієнт бачить яскраві предмети з чіткими контурами і не розрізняє об’єкти зі слабкою кольоровою гамою. У багатьох хворих відзначається порушення колірного зору за типом червоно-зеленої дісхромазіі, при якій людина бачить світло-зелений колір як темно-червоний. У той же час, деякі пацієнти не відзначають зміни сприйняття кольорової гами.
У початковій фазі захворювання не змінюються межі периферичного зору, при прогресуванні центральні скотоми розширюються, що призводить до повної сліпоти. Одночасно з появою випадання центрального зору зменшується його гострота. У завершальній стадії хвороби Штаргардта зоровий нерв атрофується. Людина повністю втрачає зір. Чи не спостерігається змін інших органів, як в початковій, так і в термінальній стадії захворювання.

Дистрофія сітківки
Діагностика
Захворювання починається в дитячому віці – це один з головних ознак для диференціальної діагностики. За допомогою офтальмоскопії виявляється широке кільце зниженою пігментації, яке оточує темний центр. Навколо блідого кільця відзначається наступне кільце гіперпігментірованних клітин. Картина нагадує «бичаче око» або «ковану бронзу». Фовеолярний рефлекс негативний. Макулярное піднесення не визначається. При огляді жовтої плями відзначаються жовтувато-білі плями різної величини і конфігурації. Згодом кордону включень розмиваються, плями набувають сірого відтінку або повністю зникають.
Під час проведення периметрії при хворобі Штангардта відзначаються позитивні або негативні (пацієнт їх не відчуває) центральні скотоми. При центральній формі захворювання розвивається червоно-зелена дейтеранопія. Для периферичної форми не характерно порушення колірного сприйняття. Просторова контрастна чутливість змінюється по всьому діапазону: відсутня в області високих частот (в центральній ділянці до 6-10 градусів) і знижується в області середніх частот.
У початковій стадії захворювання відзначається зниження показників макулярної електрографія при центральній формі дистрофії. При подальшому прогресуванні електричні потенціали не реєструються. При розташуванні дистрофії по середньої периферійній зоні в початковій стадії відзначається нормальна електрографія і електроокулографія. Потім значення колбочкових і паличкових компонентів елетроретінографіі знижуються до субнормальрних. Захворювання протікає безсимптомно – без порушення гостроти зору і сприйняття кольору. Межі поля зору знаходяться в межах норми. Незначно знижена темновая адаптація.

Хвороба Штаргардта. Етіопатогенетичні особливості
За допомогою флуоресцентної ангіографії на тлі «бичачого ока» не виявляються зони гіпофлуоресценціі, проглядаються капіляри, «мовчить» або «темна» судинна оболонка. У зонах атрофії помітні гіперфлуоресцірующіе ділянки клітин пігментного епітелію сітківки. Гістологічне дослідження в центральній зоні очного дна визначає підвищену кількість пігменту – ліпофусцину. Відзначається комбінація гіпертрофованих і атрофованих клітин пігментного епітелію.
Молекулярно-генетичний аналіз дозволяє помітити мутацію гена до початку проявів хвороби. Щоб виявити заміну нуклеотидів, проводиться ПЛР в режимі реального часу при використанні декількох ДНК-зондів – «молекулярних маяків».
Диференціальна діагностика хвороби Штаргардта здійснюється з набутими лікарськими дистрофії:
- плямами сітківки Кандор,
- сімейними друзами,
- ювенільний ретіношізіса,
- домінантною прогресуючої фовеальній,
- колбочковой,
- колбочно-палочковой і палички-колбочковой дистрофією.
Лікування
Етіологічного лікування немає. Як загальне допоміжного лікування застосовуються парабульбарно ін’єкцій таурину і антиоксидантів, введення судинорозширювальних засобів (пентоксифілін, нікотинова кислота), стероїдних препаратів. Проводиться вітамінотерапія для зміцнення судин і поліпшення кровопостачання (віт. Групи В, А, С, Е). Показані фізіотерапевтичні методи лікування: лікарський електрофорез, ультразвук, лазерстімуляція сітківки. Застосовується методика реваскуляризації сітківки шляхом трансплантації в зону жовтої плями пучка м’язових волокон. Розробляється патогенетична регенераційні офтальмологічна технологія аутологічної тканинної терапії за допомогою стовбурових клітин жирової тканини пацієнта.
Хвороба Штаргардта починається в ранньому віці і швидко призводить до інвалідності по зору. У рідкісних випадках, при домінантному типі успадкування, зір падає повільно. Пацієнтам рекомендується спостереження офтальмолога, прийом вітамінних комплексів і носіння сонцезахисних окулярів.
Читати по темі: Розрив сітківки




- Item 1
- Item 2
-

Ці 10 засобів для здоров’я нігтів є у кожній домівці
Якщо ви вважаєте, що для догляду за нігтями потрібні лише дорогі профе...
-

Ключ до довголіття: як швидкість ходьби впливає на тривалість життя
Іноді навіть невеликі зміни в способі життя можуть мати значний вплив ...
-

Цукор уповільнює розвиток мозку і розумових здібностей дитини
Діти найбільше споживають доданий цукор, навіть незважаючи на те, що д...
-

Які якості найбільше цінує перукар в поведінці свого клієнта
Гарні відносини з перукарем — це важлива умова. Розумні жінки прекрасн...
-

Зміна кольору нальоту на язиці може говорити про захворювання
Одна з найбільш поширених причин зміни кольору нальоту на язиці – хрон...
-

Вчені розповіли, чи дійсно кава гальмує ріст дітей
Однією з причин, чому каву зазвичай не дають дітям, є широко поширена ...
-

Найважливіші вітаміни для краси шкіри жінок будь-якого віку
Густе і блискуче волосся, гладка і пружна шкіра, акуратний манікюр — в...
-

Названий спосіб приготування бутерброда для довголіття
Вважається, що бутерброди – не найкорисніша їжа. Причиною тому – велик...

Як позбутися від клубка в горлі: кращі поради від медиків
У стресових ситуаціях, при захворюванні на ангіну, а також в інших випадках у кожного з нас може з'явитися відчуття клубка...


Що таке ВСД?
14.03.2024
Як лікувати шпору п’яти: сучасні методи
15.05.2023

Причини та симптоми панічних атак. Що робити
25.04.2023
Коронавірус у легкій формі: симптоми та схема лікування
Ознаки коронавірусу у легкій формі Легким перебігом ковіда, згідно з даними Всесвітньої організації охорони здоров'я, вважається наявність загальних клінічних проявів захворювання без симптомів запалення легеневої тканини (вірусної пневмонії) та гіпоксії (нестачі кисню в крові). До перших симптомів коронавірусу в легкій...

Вакцина від коронавірусу: протипоказання
Основні рекомендації вже неодноразово розглядалися та приймалися відповідними рішеннями, але є уточненими, оновленими та мають відповідний термін дії. Так, при...

Симптоми омікрон-штаму: 7 ознак того, що у вас може бути новий вид коронавірусу
Світ переполошили звістки про новий штам коронавірусу, який фахівці охрестили «омікроном» за однією з літер грецького алфавіту. Зараз вчені зосереджені...